|
E-CRISP Design of CRISPR constructs |
|
| Design | Evaluation | MultiCRISP | CLD | GenomeCRISPR | Help | Links |
Check out our new CRISPR Library Designer (CLD): batch design of sgRNA libraries Download the dockerized version now at CLD on Github |
|
Privacy |
|
| Further information about our Data Protection Policy can be found here: Privacy Policy | |
Contact |
|
|
Florian Heigwer, Marco Breinig, Tianzuo Zhan, Michael Boutros E-mail: crispr@dkfz.de |
|
Former Contributors |
|
|
Programming: Grainne Kerr, Oliver Dreier, Johanna Kratzer, Pauline Burkhardt, Alexander Mattausch, Pelz Oliver Ideas: Marco Breinig, Tianzuo Zhan, Jan Winter, Dirk Brüggemann |
|
How to cite |
|
| Heigwer, F. , Kerr, G. & Boutros, M. E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122-123 (2014). | |
Summary: |
|
|
E-CRISP is an online tool to design and evaluate Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) E-CRISP has been optimized using fast and accurate algorithms to design CRISPR gRNA sequences to target any nucleotide sequence ranging from single exons to entire genomes. Special emphasis in the design process has been given to usability in experimental applications. E-CRISP not only checks for target specificity of the putative designs but also assesses their genomic context (e.g. exons, transcripts, CpG islands). |
|
Input: |
|
| The input options are divided into 6 sections, each dealing with a different aspect of the design process |
| Section | Aspect | ||||||||||||
| Organism | The organism the designs should be created for. The databases are pre-built for each organism. The genome release is indicated in the dropdown menu. | ||||||||||||
| Target Sequence | The sequence the CRISPR should be designed to target. Either enter an Ensembl ID, a gene symbol or a sequence in fasta format. If a fasta sequence is given the locus can be stated in the header in the form of "chrom:X:1..1000 ", if your sequence originates from the first 1000 bases of the X chromosome. If this location is not be stated in the header (the text after ">") the program does not check the genomic context. | ||||||||||||
| Design Purpose |
In this section, the user can specify the experimental purpose of the CRISPR. Depending on the purpose, different regions of the input sequence will be targeted. Purposes included, knock-out experiments, N-Terminal tagging, C-Terminal tagging, CRISPRi and CRISPRa.
Further many more specific parameters can be tuned there. Such as favourable GC-content or PAMs.
|
||||||||||||
| Gene annotation filtering | In this section, the user can filter the output results, based on gene annotation information. For example, all results which do not target an exon can be excluded from the output, or the user can specify which exon to target. | ||||||||||||
| Off-target Analysis | In this section, the user can specify parameters to search for off-target effects (regions where the design targets outside of input query sequence) | ||||||||||||
| Output | In this section, the user can specify what output files are produced. If the user expects a lot of CRISPR designs to be return (e.g. inputting a large number of sequences at once), the user can switch off producing an image and an html output table. |
Output: |
|
A summary of the design process.
|
|
| A html table is returned, where each row indicates a CRISPR alignment. |
| Column | Meaning |
| Name | The ID of the CRISPR. This is of the form: ID of the input sequence_randomNumber_random_Numer. |
| Nucleotide Sequence | The sgRNA target sequence |
| SAE Score | S: Specificity score A: Annotation score E: Efficiency score. See the table below for more information. |
| Target | The gene that is targeted by this gRNA. If a fasta sequence is given as input with no chromosome location information, E-CRISP cannot search the annotation databases, and no target gene will be returned. |
| Match String | A coloured match screen, which indicates at a glance how good the alignment is: A green "M" for a match. A "X" for a mismatch, an "I" for an insertion in the gRNA. |
| Number of Hits | The number of locations this CRISPR design targets, or, the number of times this CRISPR appears in the output table (one for each target). |
|
A genome browser image of the CRISPR designs in their genomic context. This allows the user to visually inspect where the CRISPR in the input sequence. Off-targets (where the CRISPR targets outside of the input sequence) are not shown in this image.
There is also an option to output a gff file (http://www.ensembl.org/info/website/upload/gff.html) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The output table is available as tab-delimited file *.tab or Excel compatible file *.xls. And contains the following columns: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Column | Meaning |
| Name | target site ID |
| Length | target length |
| Start | target start with respect to the input sequence (gene -500 if a gene name was the input) |
| End | target start with respect to the input sequence (gene -500 if a gene name was the input) |
| Strand | strand it will target |
| Nucleotide sequence | target site nucleotide composition of the form target_PAM |
| Gene Name | ID::GENE |
| Transcripts | ENSEMBL transcript Ids overlapping with the target site |
| Transcript:: Exon | ENSEMBL transcript::exon Ids overlapping with the target site |
| Number of Cpg Islands hit | Number of Cpg Islands overlapping with the target site |
| Sequence around the cutside | if it is chosen to save a recombination matrix ist sequence is here |
| %A %C %T %G | nucleotide compositions in per cent |
| S-Score | specificity score |
| A-Score | annotation score |
| E-Score | efficacy score |
| percent of total transcripts hit | per cent of transcripts of the targeted gene being hit by that putative sgRNA |
| Target | target genes by remapping the target site |
| Match-start | alignment start with respect to the estimate target gene |
| Match-end | alignment end with respect to the estimate target gene |
| Matchstring | alignment representation "M" for match "X" for mismatch "I" for insertion "D" for deletion |
| Editdistance | estimated edit distance of the alignment (X+I+D) |
| Number of Hits | estimate number of target sites in the respective genome with the off-target parameters specified |
| Direction | strandedness of the target alignment |
| CDS_score | start with 0. for every CDS of every transcript the target site ovelaps with add 5/CDS_number |
| Exon_Score | start with 0. for every exon of every transcript the target site ovelaps with add 5/exon_number |
| seed_GC | GC content in per cent of the 8 PAM proximal basepairs |
| Doench_Score | Efficacy score as introduced by Doench et al. 2014 Nat. Biotech. |
| Xu_score | Efficacy score as introduced by Xu et al. 2015 Gen.Res. |
| Chromosome | chromosome the sgRNA targets |
| Genomic start | genomic location on that chromosome the sgRNA is targeting |
| Genomic End | genomic location on that chromosome the sgRNA is targeting |
Scoring: All scores given are normalized to 100 % reachable score
In addition the scores of Doench et al. and Xu et al. are given in the output table.
| Specificity Score (S-score) | Annotation Score (A-score) | Efficacy Score (E-score) |
|
Start with 100 for every off-target substract (20-mismatches)/iteration |
Start with zero For every hit exon add 5/exon count For every hit CpG Island subtract 1 For every start codon hit add 1 For every stop codon hit add 1 For every CDS hit add 5/CDS count For every gene hit add 1 |
Add 1 if the the last 6 bp have a CG content higher then 70 % Subtract 1 if the entire sequence has GC content > 80 % Add 1 if sequence is preceded by a G Add 1 if there are GG in front of the target sequence (opposite the PAM) Add micro -homology score (is higher when sequence tends to give out of frame deletions) |
Frequently asked questions:
Why do I have to choose the organism for my design?
How does E-CRISP annotate my sequence?
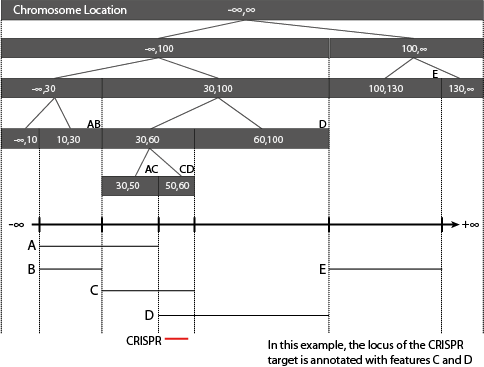
How does E-CRISP identify off-targets?
Why do I have a choice against which sequence to check for off-targets?
How do I identify if my CRISPR design has any off-targets from the output table?
What is a secondary off-target?
I only get a chromosome name in the target column of the output table - why?
Why is the minimum/maximum guide RNA length after PAM 20?
How does E-CRISP work?
Where is the information about the organisms taken from?
Change Log:
| 2 Jul 2019, version 5.4 | 1. An option to exclude homopolymer stretches of nucleotides has been added. 2. A bug, whereby homopolymers were only searched in forward direction has been removed. 3. Stringency levels were adapted such that strict parameters are less strict and all respect the homopolymer exclusion 4. Homo sapiens became the default organism. |
| 12 Jul 2016, version 5.1 | 1. Again new organisms including Lifestock, crops, funghi and bacteria have been added to E-CRISP 2. All other databses were updated to the newest version available in ensembl. 3. TSS, start codons and stop codons can be vizualized in the results 4. Scoring schemes of Doench et al. and Xu et al. added to the analysis. 5. Genomic location added to the output 6. Excel support added to the output 7. Output image beatified 8. fixed some minor bugs regarding the strandedness of genes and thus tagging results 9. implmented,tested and fixed rulesets for CRISPRa and CRISPRi 10. fixed flexible PAM input |
| 22 Jan 2015, version 4.2 | 1. Many new organisms including Lifestock, crops, funghi and bacteria have been added to E-CRISP 2. All other databses were updated to the newest version available in ensembl. 3. Annotations have to overlap with the pam and not only some part of the sgRNA target 4. Targets can be identified in sequences which do not originate from the organism selected. 5. CpG islands are again properly shown in the result image |
| 01 August 2014, version 4.0 | E-CRISP has been reworked to inlcude the latest scientific results of the last months: 1. The following organisms hav been added: Toxoplasma gondii GT1 (ToxoDB-10.0) Gasterosteus aculeatus (Three-spined stickleback, BROADS1.75) Populus trichocarpa (Black cottonwood, JGI2.0.21) Sus scrofa (Pig, Sscrofa10.2.75) 2. A new more intuitive scoring system, devided into Specificity, Annotation and Efficiency score has been implemented. Design results are sorted by Specificity, then Annotation and then efficiency 3. Three new default options have been added guiding you fastly to the most wanted results. For further details visit the help pages and scroll down to the schoring scheme. 4. Off-target checks are now much more precise, because the PAM region (NAG or NGG) now is truely ambigous. An off-target is searched without the PAM but only considered valid if any PAM is present. |
| 26 May 2014, version 3.1 | We are happy to announce a further major update to our E-CRISP web service. Many new organisms have been added together with big changes in the web front end. Hence you will find the new forum and many other new things here in the new BETA version 3.1. |
| 14 April 2014, version 3.0.2 | In this minor update different default values for de-novo sgRNA design have been implemented, allowing for more designs to be found. |
| 01 April 2014, version 3.0.1 | The following organisms have been added to E-CRISP:
Zea mays Ustilago hordei Toxoplasma gondii ME49 Gasterosteus aculeatus Populus trichocarpa |
| 20 March 2014, version 3 | A new version of E-CRISP has been released (version 3.0). It includes more off-target search options and we implemented speed improvements to enable the design of sgRNAs against up to 200 genes in parallel. |
References:
Boutros lab, E-CRISP-Version 5.4 For suggestions please contact us at crispr@dkfz.de |